Terapia celular en neuropatologías
Fisiopatología integrativa y terapia
Descripción
Introducción
Nuestro grupo trabaja de forma multidisciplinar para comprender, diagnosticar y tratar algunas encefalopatías epilépticas y del desarrollo especialmente graves: los síndromes STXBP1, de West y de Dravet. Afectan a niños desde los pocos meses de vida y se caracterizan por epilepsia farmacorresistente, disfunción neurocognitiva severa y alteraciones motoras debidas principalmente a una disfunción en la actividad de las interneuronas.
Mediante aproximaciones experimentales que combinan terapia génica y celular en modelos animales, electrofisiología, conducta, neurobiología molecular, bioinformática y biología de sistemas, buscamos desarrollar estrategias terapéuticas innovadoras que mejoren la calidad de vida de los pacientes, comprender mejor estas patologías y sus comorbilidades, y avanzar hacia una medicina personalizada.
Líneas de trabajo
• Terapia celular mediante precursores neuronales GABAérgicos para las encefalopatías epilépticas y del desarrollo
• Terapia génica mediante oligos antisentido (ASOs) para el síndrome STXBP1
• Análisis multiómicos de encefalopatías epilépticas y del desarrollo
• Implicación del canal Nav1.1 en los trastornos mentales durante la adolescencia
Terapia celular para los síndromes STXBP1, de West y de Dravet
Nuestro grupo desarrolla estrategias de terapia celular orientadas a restaurar el desequilibrio existente entre la actividad excitatoria e inhibitoria en los síndromes STXBP1, de West y de Dravet, lo que conduce a crisis epilépticas, discapacidad intelectual, trastornos del movimiento, problemas conductuales y alteraciones del sueño. La disfunción de las interneuronas GABAérgicas es un componente clave en ambas patologías y un factor central en la aparición de crisis epilépticas resistentes a fármacos. Nuestra línea de trabajo se basa en el trasplante de precursores neuronales derivados de la eminencia ganglionar medial, que durante el desarrollo genera de forma natural interneuronas funcionales. El aislamiento, o generación a partir de células madre pluripotentes inducidas (IPSCs), y su posterior trasplante en modelos murinos nos permiten evaluar la integración sináptica de estos precursores, su contribución a la normalización de la actividad inhibitoria y su capacidad para reducir las crisis epilépticas, así como el rescate de los déficits cognitivos, conductuales y alteraciones del sueño. Los resultados preliminares muestran mejoras en la actividad inhibitoria, reducción de las crisis, y beneficios en aspectos cognitivos y conductuales. Esta línea se articula y financia gracias a la colaboración de las asociaciones nacionales de enfermos del síndrome STXBP1 y Apoyo Dravet, así como la Fundación Ramón Areces.
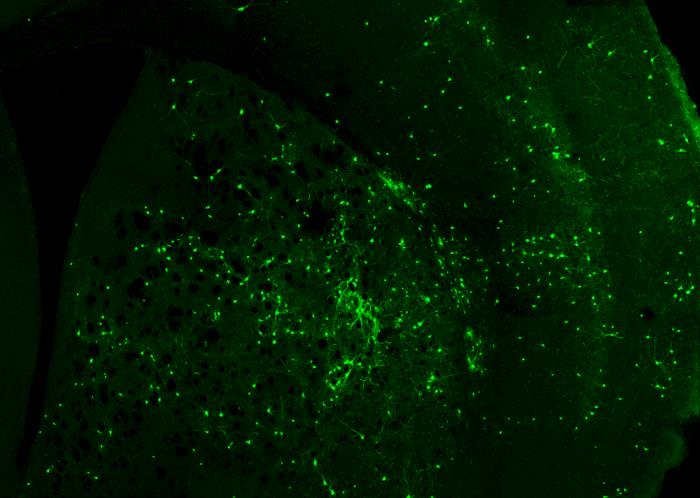
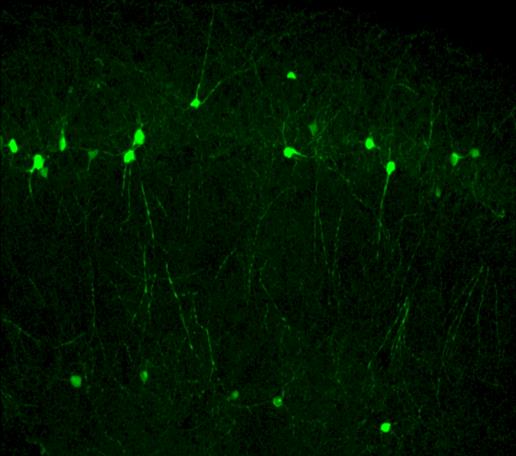
Terapia génica mediante oligos antisentido para el síndrome STXBP1
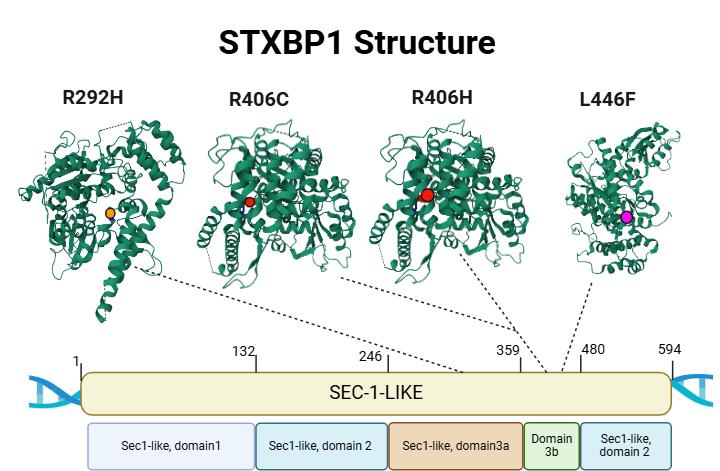
El síndrome STXBP1 está causado por mutaciones en el gen del mismo nombre, que reducen drásticamente los niveles de la proteína Munc18-1 que codifica. Esta proteína es un componente esencial del complejo SNARE, que constituye la maquinaria de exocitosis de las vesículas sinápticas. Por ello se le califica como una SNAREopatía. Basándonos en esta fisiopatología, diseñamos ASOs capaces de unirse a regiones UTR del pre-mRNA para aumentar su estabilidad y evitar su degradación temprana, con lo que logramos incrementar su traducción y los niveles de Munc18-1. Asimismo, estamos desarrollando ASOs personalizados dirigidos específicamente a transcritos mutados, que permiten la eliminación selectiva de mRNAs aberrantes que dan lugar a proteínas truncadas aberrantes que favorecen la agregación proteica y la disfunción celular.
La liberación selectiva de los ASOs en el cerebro la estamos desarrollando mediante una vehiculización basada en vesículas extracelulares, en colaboración con el grupo de la Dra. Lydia Álvarez Erviti del CIBIR. Mediante la ingeniería de vesículas exosomales que expresan el péptido RVG (rabies virus glycoprotein), logramos dotarlas de tropismo neuronal y favorecer su transporte transendotelial a través de la barrera hematoencefálica. Esto permite la administración sistémica (intravenosa o intranasal) de ASOs encapsulados, evitando procedimientos invasivos como la inyección intraventricular repetida. La evaluación funcional de estas plataformas se lleva a cabo mediante líneas celulares neurales (Neuro-2a), modelos murinos heterocigotos STXBP1+/− y cultivos ex vivo de neuronas corticales e hipocampales. Esta línea se financia gracias a la colaboración de la asociacion nacional de enfermos del síndrome STXBP1, la Unión Europea (Proyectos ERDERA-JTC 2025; AC25/00053) y la Junta de Andalucía (Proyectos de investigación e innovación de colaboración público-privada 2024, de la Consejería de Salud y Familia, PIP-0113-2024).
Análisis multiómicos de encefalopatías epilépticas y del desarrollo
En colaboración con el Dr. Francisco J. Esteban de la Universidad de Jaén y los Neuropediatras Rocío Calvo (Hospital Regional Universitario de Málaga) y Gil-Nagel (Hospital Ruber Internacional), estamos analizando mediante herramientas bioinformáticas y de biología de sistemas el genoma, transcriptoma y metaboloma de modelos animales y pacientes STXBP1 y Dravet. Este enfoque combina herramientas de análisis de variantes a partir de datos de genoma completo, datos clínicos, estimación de riesgo poligénico, identificación de genes reguladores y reconstrucción de redes de expresión diferencial basadas en datos RNA-seq. A través de modelos estadísticos y algoritmos de inteligencia artificial aplicados al análisis multimodal, buscamos definir relaciones genotipo-fenotipo con mayor precisión, identificar biomarcadores de diagnóstico y prognosis, y desarrollar modelos estratificados que faciliten la medicina personalizada para un tratamiento antiepiléptico más certero y eficaz. Esta línea se financia gracias a la colaboración de la asociación nacional de enfermos del síndrome STXBP1, la Fundación Alicia Koplowitz y la Junta de Andalucía (Proyectos de investigación e innovación de colaboración público-privada 2024, de la Consejería de Salud y Familia, PIP-0113-2024).
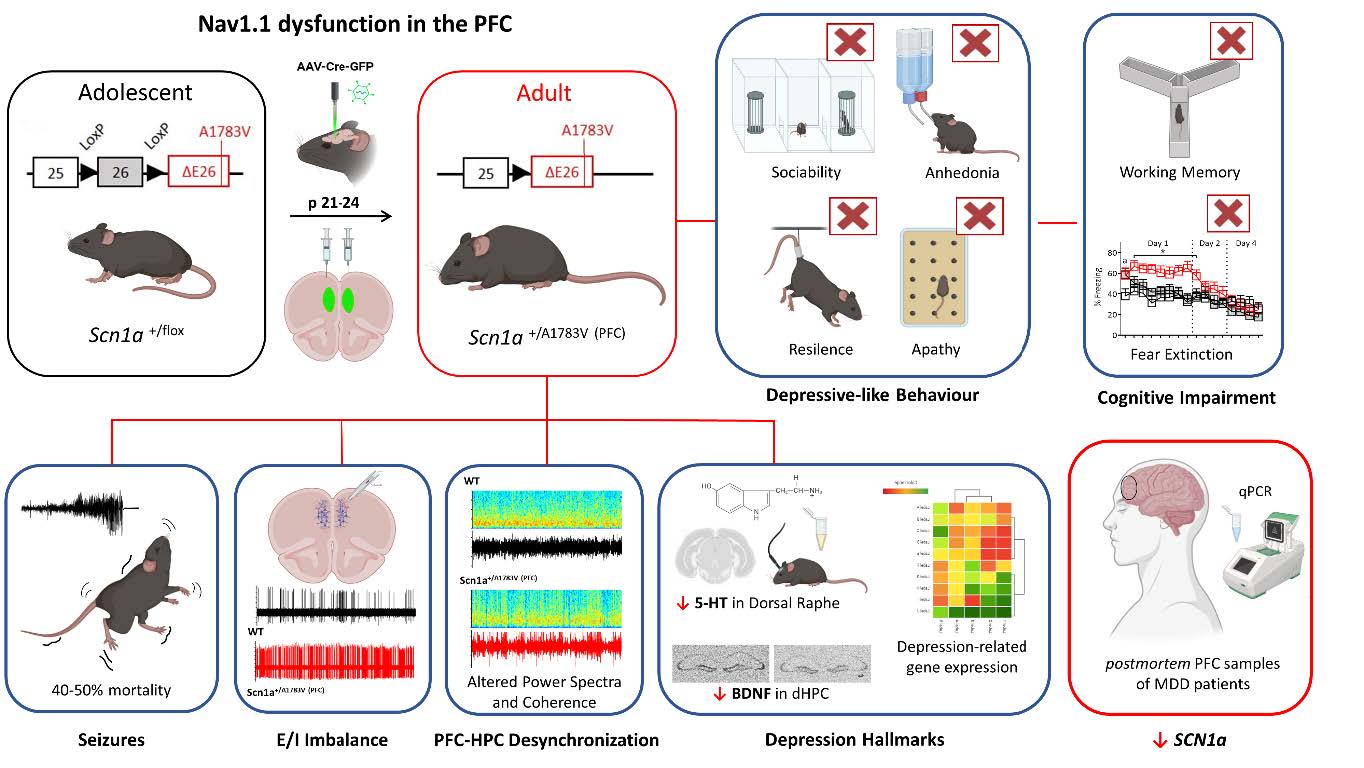
Implicación del canal Nav1.1 en los trastornos mentales durante la adolescencia
Mutaciones en el gen SCN1A, que codifica para el canal de sodio dependiente de voltaje Nav1.1, causan el síndrome de Dravet. Pero la función de Nav1.1 es también muy importante en otras neuropatopatologías graves como el Alzheimer o la depresión. En esta línea de trabajo estudiamos el papel que juega Nav1.1 en la corteza prefrontal, última región del cerebro en madurar y que es crítica para la toma de decisiones, la regulación emocional y la conducta social. Alteraciones en las interneuronas de esta región, en donde se expresa Nav1.1, se han asociado a numerosas patologías neuropsiquiátricas. Recientemente, hemos demostrado que la disfunción de Nav1.1 en la corteza prefrontal durante la adolescencia causa, en paralelo a la epilepsia, déficits cognitivos y fenotipos depresivos severos en la edad adulta. La disfunción de Nav1.1 en interneuronas GABAérgicas, particularmente en los subtipos PV+ y SST+, altera la dinámica inhibidora cortical y contribuye a fenotipos conductuales diferencialmente asociados a rasgos depresivos, autistas, hiperactividad o déficits cognitivo-afectivos. Esto demuestra el papel crítico del canal Nav1.1 en la maduración y estabilidad de los circuitos prefrontales y sugiere su relevancia como nueva diana terapéutica.
Esta línea está financiada por el Ministerio de Ciencia, Innovación y Universidades; Agencia Estatal de Investigación; Proyectos de Generación de Conocimiento (PID2024-162613OB-I00 y PID2021-127044OB-I00).
Miembros actuales



Contacto
Email: contacto@cabimer.es
Web: https://cabimer.sombradoble.es