Cell therapy for neuropathologies
Integrative Pathophysiology and Therapies
Description
Introduction
Our group takes a multidisciplinary approach to understand, diagnose, and develop treatments for several particularly severe developmental and epileptic encephalopathies, including STXBP1 syndrome, West syndrome, and Dravet syndrome. These disorders affect children from the first months of life and are characterized by pharmacoresistant epilepsy, severe neurocognitive impairment, and motor abnormalities, largely resulting from dysfunction in inhibitory interneuron activity.
By combining gene and cell therapy approaches in animal models with electrophysiology, behavioral analysis, molecular neurobiology, bioinformatics, and systems biology, we aim to develop innovative therapeutic strategies that improve patients’ quality of life. At the same time, we seek to deepen our understanding of these disorders and their comorbidities, ultimately contributing to the development of personalized therapeutic approaches.
Research Areas
• Cell therapy using GABAergic neuronal precursors for developmental and epileptic encephalopathies
• Gene therapy using antisense oligonucleotides (ASOs) for STXBP1 syndrome
• Multi-omics analyses of developmental and epileptic encephalopathies
• Role of the Nav1.1 channel in psychiatric disorders during adolescence
Cell Therapy for STXBP1, West, and Dravet Syndromes
A major focus of our research is the development of cell-based therapeutic strategies aimed at restoring the imbalance between excitatory and inhibitory neuronal activity observed in STXBP1, West, and Dravet syndromes. This imbalance contributes to epileptic seizures, intellectual disability, movement disorders, behavioral abnormalities, and sleep disturbances. Dysfunction of GABAergic interneurons represents a key pathological mechanism in these disorders and plays a central role in the emergence of drug-resistant seizures. Our work explores the transplantation of neuronal precursors derived from the medial ganglionic eminence (MGE), a developmental structure that naturally generates functional cortical interneurons. These precursors can be isolated directly or generated from induced pluripotent stem cells (iPSCs). Their subsequent transplantation into murine models allows us to evaluate their synaptic integration, their contribution to restoring inhibitory activity, and their ability to reduce epileptic seizures, as well as to rescue cognitive and behavioral deficits and sleep alterations. Preliminary results indicate improvements in inhibitory network activity, reduced seizure frequency, and beneficial effects on cognitive and behavioral outcomes. This research line is developed and funded through collaboration with the national patient associations for STXBP1 syndrome and Apoyo Dravet, as well as the Ramón Areces Foundation.
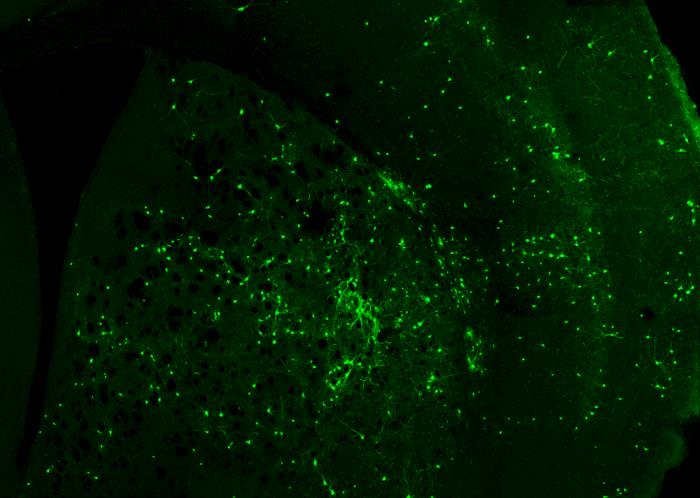
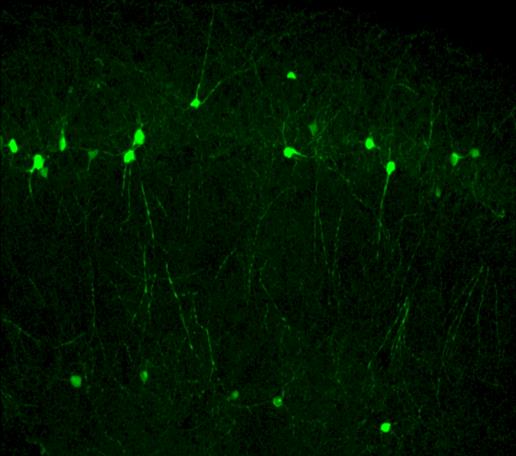
Gene Therapy Using Antisense Oligonucleotides for STXBP1 Syndrome
STXBP1 syndrome is caused by mutations in the STXBP1 gene, which drastically reduce the levels of the protein Munc18-1 that it encodes. This protein is an essential component of the SNARE complex, the molecular machinery responsible for synaptic vesicle exocytosis. For this reason, STXBP1 syndrome is often classified as a SNAREopathy. Based on this pathophysiological mechanism, we design antisense oligonucleotides (ASOs) capable of binding to untranslated regions (UTRs) of the pre-mRNA, increasing transcript stability and preventing early degradation. This approach enhances translation and ultimately increases the levels of Munc18-1 protein. In parallel, we are developing personalized ASOs specifically targeting mutated transcripts, enabling the selective elimination of aberrant mRNAs that produce truncated proteins that can promote protein aggregation and contribute to cellular dysfunction. To achieve selective delivery of ASOs to the brain, we are developing a delivery vehicle based on extracellular vesicles, in collaboration with the group of Dr. Lydia Álvarez Erviti at CIBIR. Through the engineering of exosomal vesicles expressing the RVG peptide (rabies virus glycoprotein), we confer neuronal tropism and promote transendothelial transport across the blood–brain barrier. This strategy enables the systemic administration of encapsulated ASOs (intravenous or intranasal), avoiding invasive procedures such as repeated intraventricular injections. The functional evaluation of these delivery system is carried out using neural cell lines (Neuro2a), STXBP1+/− heterozygous mouse models, and ex vivo cultures of cortical and hippocampal neurons. This research line is supported by the Spanish STXBP1 patient association, the European Union (ERDERA-JTC 2025; AC25/00053), and the Regional Government of Andalusia through the Public–Private Collaborative Research and Innovation Program (2024), Ministry of Health and Families (PIP-0113-2024).
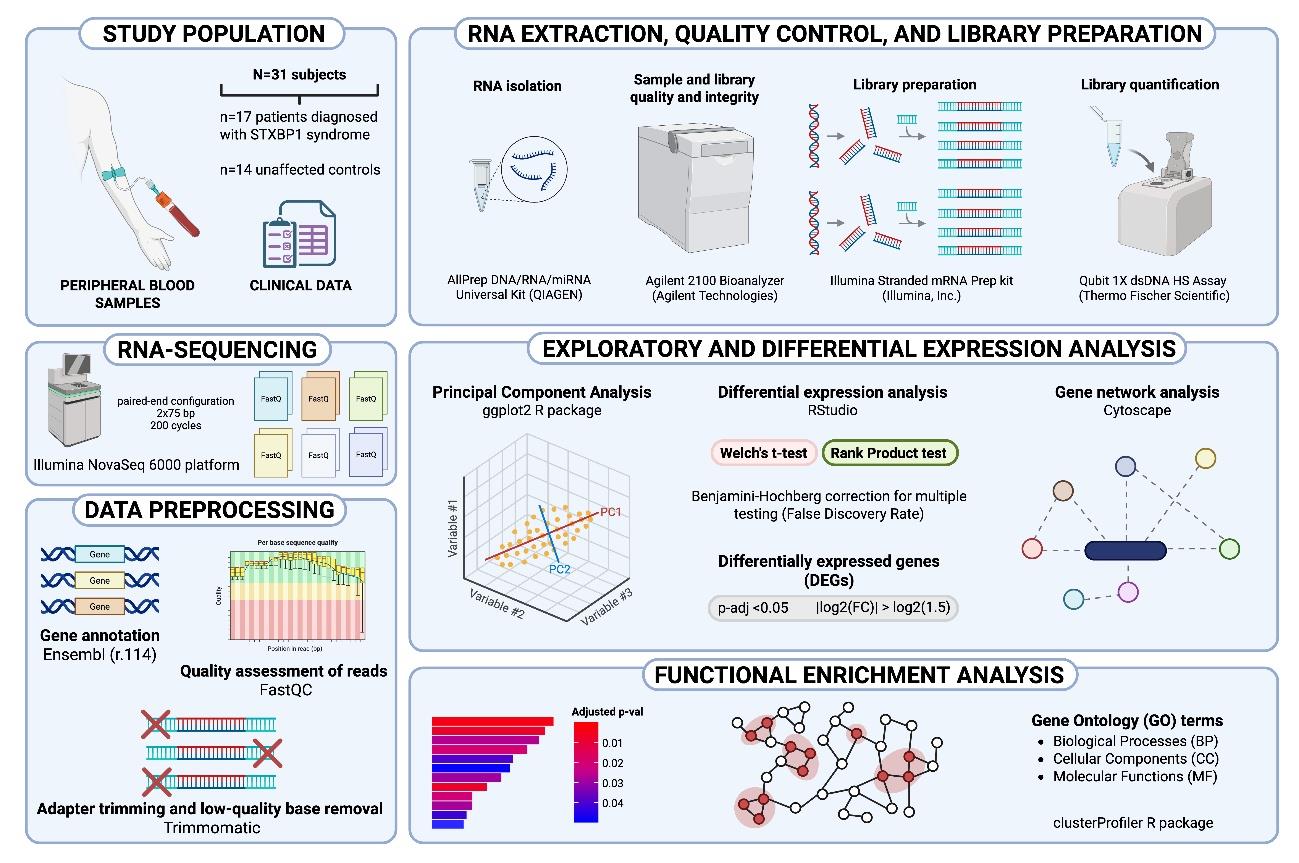
Multi-Omics Analysis of Developmental and Epileptic Encephalopathies
In collaboration with Dr. Francisco J. Esteban (University of Jaén) and pediatric neurologists Dr. Rocío Calvo (Regional University Hospital of Málaga) and Dr. Gil-Nagel (Ruber International Hospital), we are applying bioinformatics and systems biology approaches to analyze the genome, transcriptome, and metabolome of both animal models and patients with STXBP1 and Dravet syndromes. This integrative approach combines whole-genome variant analysis, clinical data integration, polygenic risk estimation, regulatory gene identification, and reconstruction of differential gene-expression networks based on RNA-seq data. Using statistical models and artificial intelligence algorithms applied to multimodal datasets, we aim to define genotype–phenotype relationships with greater precision, identify diagnostic and prognostic biomarkers, and develop patient stratification models that support personalized medicine approaches for more accurate and effective antiepileptic treatments. This research line is founded through the Spanish STXBP1 patient association, the Alicia Koplowitz Foundation, and the Regional Government of Andalusia through the Public–Private Collaborative Research and Innovation Program (2024), Ministry of Health and Families (PIP-0113-2024).
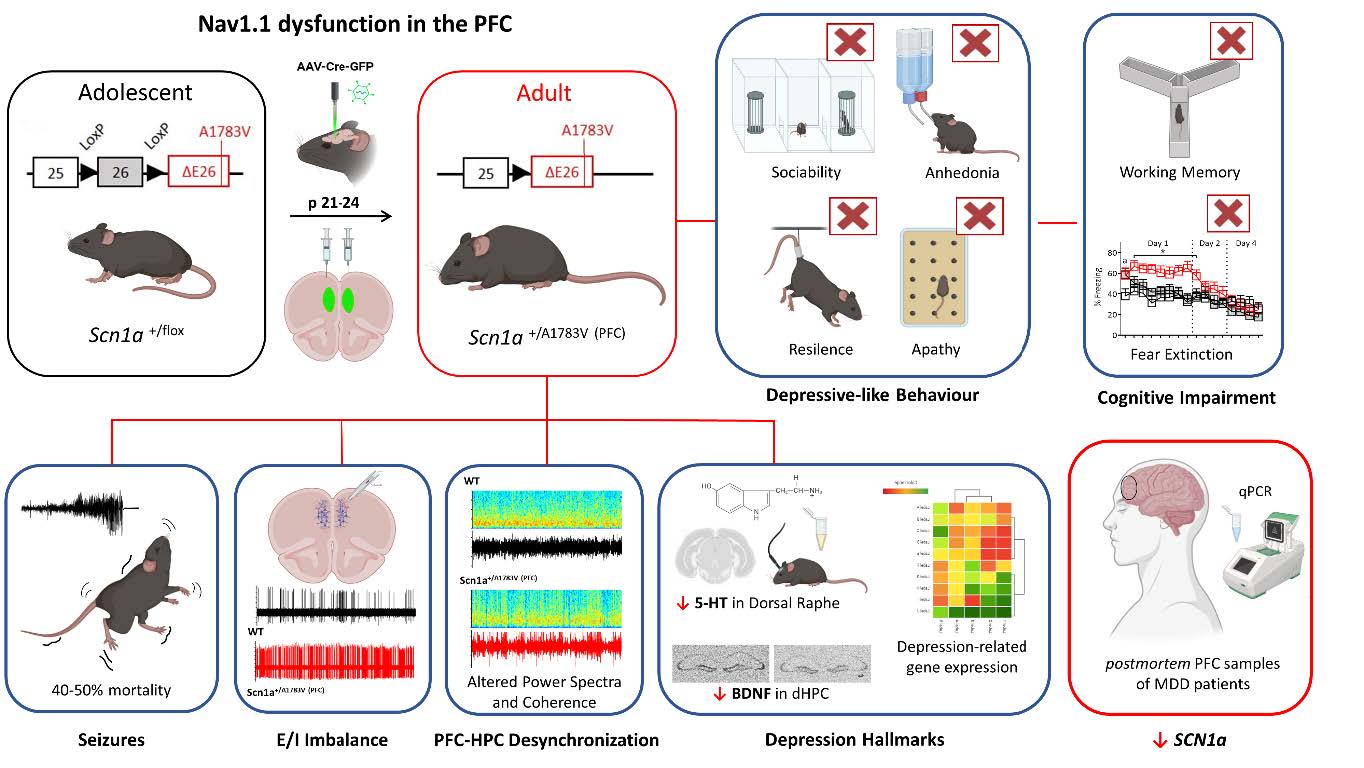
Role of the Nav1.1 Channel in Mental Disorders During Adolescence
Mutations in the SCN1A gene, which encodes the voltage-gated sodium channel Nav1.1, cause Dravet syndrome. However, Nav1.1 function is also critically involved in other severe neurological disorders, including Alzheimer’s disease and major depression. In this research line, we investigate the role of Nav1.1 in the prefrontal cortex, the last brain region to reach full maturation and a key area for decision-making, emotional regulation, and social behavior. Alterations in interneurons within this region, where Nav1.1 is highly expressed, have been associated with a wide range of neuropsychiatric disorders. Recently, we have demonstrated that Nav1.1 dysfunction in the prefrontal cortex during adolescence leads, in addition to epilepsy, to cognitive deficits and severe depressive-like phenotypes in adulthood. Disruption of Nav1.1 in GABAergic interneurons, particularly in parvalbumin-positive (PV+) and somatostatin-positive (SST+) subtypes, alters cortical inhibitory dynamics and contributes to behavioral phenotypes differentially associated with depressive traits, autism-related behaviors, hyperactivity, and cognitive-affective deficits. These findings highlight the critical role of Nav1.1 in the maturation and stability of prefrontal circuits and suggest its potential relevance as a novel therapeutic target. This research line is funded by the Spanish Ministry of Science, Innovation and Universities, the State Research Agency (Agencia Estatal de Investigación), through the Knowledge Generation Projects program (PID2024-162613OB-I00 and PID2021-127044OB-I00).